- 48小时新闻排行
- 7天新闻排行

| 2023 年 9 月 28 日,NEJM 刊载一篇“死亡报告”:一名 27 岁男性在入组某临床试验的第 6 天心脏骤停,第 8 天死亡。  论文截图 然而,很少有人知道,在这篇极尽科学与前沿的论文背后,是一名哥哥 5 年的努力。 为了给身患绝症的弟弟开发一款独一无二的“新药”,他集齐全球顶尖医学专家,拿到巨额投资,开展了“仅有一名受试者”的临床试验。 身患罕见病的弟弟 时间回到 2018 年。某天,还在哈佛大学商学院攻读研究生的理查德(Richard Horgan)偶然看到一则这样的新闻。 一名 6 岁女孩罹患致命性神经系统罕见病,保守治疗效果甚微,生命危在旦夕。在她的家人的不懈努力之下,来自顶级儿童医院的医学专家仅用 10 个月时间,就为女孩开发出了“定制化”治疗药物,并迅速改善了女孩的症状。 这一奇迹般的事件一时间[1]被全美媒体争相报道,女孩的治疗经过也以论文的形式发表在 NEJM 上[2]。 在普通人眼中,这可能只是一个茶余饭后的感人故事,然而,这个新闻对理查德来说意义非凡——他的亲弟弟特里(Terry Horgan)也是一名罕见病患者。 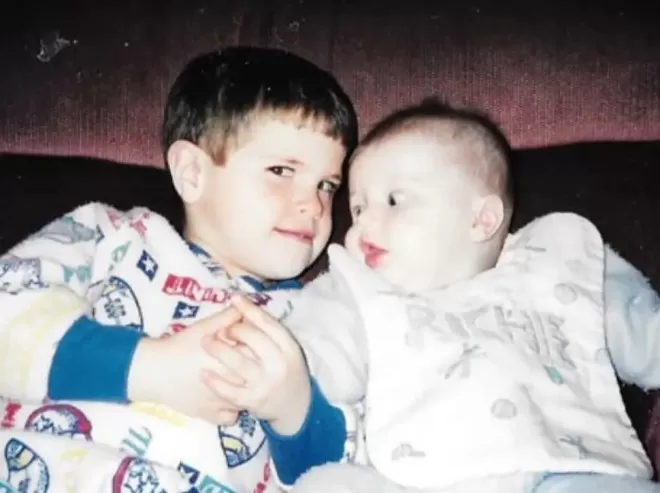 5 岁时,特里被诊断患有杜氏肌营养不良(DMD)。这是一种罕见的遗传性肌肉疾病,在婴幼儿中发病率有 1/3300[3],且致残率极高。典型的 DMD 可早在 2~3 岁时即发病,到了 12~13 岁时,患者就只能依赖轮椅生活,平均寿命不足 20 岁。 导致 DMD 的原因在于抗肌萎缩蛋白基因的失活。在肌肉中,抗肌萎缩蛋白具有保护肌纤维细胞膜、预防运动后肌肉疲劳、阻止钙离子病理性流入肌纤维的作用,而 DMD 患者中抗肌萎缩蛋白的严重缺乏导致了肌纤维的严重变性[4],进而诱发肌肉组织的结构紊乱和功能丧失。 尽管皮质类固醇可以在一定程度上减缓 DMD 患者的肌力受损和病情恶化,但大多数患者的病情仍进展迅速,且皮质类固醇还会带来糖尿病、肥胖和情绪异常等副作用,影响患者的生活质量。更多的时候,患者只能倚赖如轮椅、气管切开等保守治疗手段。 特里也同样,他自确诊起就在服用地夫可特(一种专用于治疗 DMD 的强效皮质类固醇),但在 18 岁时,他还是丧失了行走能力,日益恶化的心肺功能更是时刻威胁着他的生命。 但特里又不太一样,因为他有一个为了弟弟倾其所有的哥哥。 基因测序寻求解法 早在 2016 年,治疗 DMD 的反义寡核苷酸药物依特立生(Eteplirsen)就已经获得了美国食品药品监督管理局(FDA)的加速批准。其他直接针对 DMD 病因的试验性治疗也陆续进入临床研究阶段。 然而,特里并无机会体验这些革命性的治疗,因为他的年龄过大,并不在这些临床研究的招募范围内。 但哥哥理查德并没有放弃。 在当时,尚未毕业的理查德已经为弟弟成立了一所非营利慈善机构 Cure Rare Disease,专注于拯救特里的生命。 当他看到了 6 岁女孩被治愈的奇迹故事时,念头只有一个:我的弟弟能否也用上这种神奇的“定制化治疗”? 理查德首先联系上了为 6 岁女孩设计“定制疗法”的医生。但医生告诉他,所谓“定制疗法”是一种反义寡核苷酸类药物,理论上不能用于特里的情况。 理查德没有放弃,他又联系了耶鲁大学一名遗传学领域专家,为特里的骨骼肌细胞进行全面的基因测序。 几周之后,一个坏消息从测序实验室传来:特里的 DMD 致病突变为抗肌萎缩蛋白启动子和外显子 1 部分缺失,这导致特里不适合目前为止所有上市或正处于临床研究的疗法。 不过,还有一个好消息,这种突变可以使用最新的基因编辑技术 CRISPR 进行基因治疗。 理查德等的就是这句话。在测序结果返回时,他已经带着一支“整装待发”的团队准备为自己的弟弟研发“定制化治疗”,这支团队云集众多顶尖研究机构的遗传病治疗专家,大部分人都是无薪受雇于 Cure Rare Disease。 筹资千万,量身定制治疗方案 团队为理查德提出的方案,是一种和 6 岁罕见病女孩思路类似的“定制化治疗”,只不过,治疗方法从反义寡核苷酸换成了 CRISPR 基因编辑。 CRISPR 是一种源于细菌的天然的核酸定位切割工具,被公认为是高效基因“剪刀”。不仅如此,某些经过特殊加工的 CRISPR 体系不会“剪掉”任何基因,反而可以定向调控基因表达[6][7],被称为“CRISPR 激活”。 理查德团队锚定了“CRISPR 激活”方案。 进一步研究发现,特里骨骼肌细胞内,合成的抗肌萎缩蛋白主要可分为三型:肌肉型(Dp427m)、皮质型(Dp427c)、和浦肯野型(Dp427p1),这三种蛋白中,只有 Dp427m 是存在严重缺陷的,另外两者则完好无损。而有资料[8]发现,Dp427c 在肌肉中可能会发挥和未突变的肌肉型抗肌萎缩蛋白相同的效果。 能否让 Dp427c 代替 Dp427m? 基于这些研究,团队决定开始冒险使用最新的“CRISPR 激活”技术,利用一种基因治疗中非常常见的载体——重组腺相关病毒(rAAV)血清型 9,在它的基因组上携带了一段特殊的 RNA 片段。 在 rAAV 输注进人体后,病毒开始陆续感染包括骨骼肌细胞在内的人体各细胞,而病毒中这段特殊 RNA 片段会“靶向性”地找到 Dp427c 启动子的位置,并激活这个原本在肌肉细胞基本不表达的类型。 这样,大量具有功能的 Dp427c 被合成,代替原有的缺陷型 Dp427m,以此改善肌肉的发育和存活情况。 那么,为什么说这是一种“定制化治疗”?这是因为,这种治疗方案需要对患者的致病基因进行测序,根据测试结果“量身打造”适合使用的药物。也就是说,对于特里极其罕见的基因突变,制造出来的药物并不能适用于其他 DMD 患者。 这也是人类首次将 CRISPR 技术引入罕见病定制化治疗[9]。 方案已经基本成熟,下一个问题是:钱。 尽管这些顶尖医学专家都是无薪受雇于理查德的非盈利机构,但并不意味着开发新的治疗方法没有成本。据团队估算,这种“定制化”CRISPR 疗法治疗一名患者的费用通常在 200~300 万美元之间。 为了弟弟的治疗,理查德开始了的筹款之路,他承诺 Cure Rare Disease 将维持起非营利机构,并会将筹得资金的一部分用于对治疗其他罕见病的药物的研发。最终筹到了 230 万美元(约合人民币 1679 万元)[10]。 只有一个人的临床试验 2022 年 8 月,FDA 授予 Cure Rare Disease 开发的 CRISPR 基因治疗疗法“研究性新药”资格,这也就意味着特里可以正式接受治疗了——不过是以临床试验的形式。 2022 年 10 月 4 日,特里注射了为他“量身定制”的搭载有 CRISPR 工具的 rAAV,输注效果良好。 似乎,一切都在往好的方向发展。 然而,注射后第二天,特里出现了室性早搏和血小板减少的迹象;与此同时,B 型钠尿肽、转氨酶、肌酸激酶的水平逐渐上升;注射后第五天,特里出现了典型的心肌炎症状;注射后第六天,特里出现了急性呼吸窘迫综合征和心力衰竭。 注射后第八天,医生宣告了特里的死亡。 随后,理查德通过 Cure Rare Disease 公布特里去世消息,并表示愿意让弟弟接受尸检解剖,以调查最终死因。 2023 年 9 月 28 日,NEJM 刊载了特里的“死亡报告”。尸检结果表明,rAAV 进入特里体内之后诱发了强烈的细胞免疫反应,导致大量免疫细胞释放炎症因子,最终导致特里死于 rAAV 相关的毛细血管渗漏综合征[11]。 实际上,作为基因治疗病毒载体的 rAAV 一直饱受安全性争议。由于腺病毒感染在自然界中过于常见(感冒的病原体之一就是腺病毒),很多人都会有针对 AAV 的抗体或交叉抗体,这导致如果对已经存在抗 AAV 抗体的患者进行基因治疗,将存在疗效丧失乃至严重免疫不良反应的风险[12]。 报告认为,特里和其他接受了相似或更高剂量 rAAV 的患者相比,出现了更严重的先天免疫反应。同时,特里接受了按公斤体重计算的药物剂量,但他因肌肉萎缩,肌肉净含量较低,可能导致药物剂量相对较高。 特里的故事震惊了基因治疗领域的研究人员,也再次为安全性问题敲响警钟。但必须承认的是,CRISPR 基因治疗依然是不少罕见病、绝症患者的最后一线希望。 正如 Cure Rare Disease 官网所写:“我们的故事并没有结束,还有成千上万的特里在等待反击的机会。”  致谢:本文经 浙江大学医学院附属第二医院神经内科副主任医师 刘功禄 专业审核 四川师范大学微生物学硕士 孙铭君 对本文亦有贡献 免责声明:本网转载的文章仅为传播更多信息之目的,本网未独立核实其内容真实性,文章也不代表本网立场。如文章侵犯了你的权利,请联系我们修改或删除。本网提供的内容,包括并不限于财经、房产类信息,仅供参考,不构成投资建议;本网内容,包括并不限于健康、保健信息,亦非专业意见、医疗建议,请另行咨询专业意见。本网联系邮箱:contact@cacnews.ca  |

特朗普猜错了 古巴被断油半年至今仍未垮台 人民靠4种替代能源苦撑
国际 16 分钟前
体育 半小时前
中国 半小时前
特朗普一边拉拢习近平一边撒钱抗中 美国政府拟加码数亿美元抢影响力
美国 1 小时前
体育 1 小时前
国际 1 小时前
娱乐 1 小时前
只花38天 那斯达克100跌到逼近修正、SpaceX蒸发1/5市值
财经 2 小时前
关注获得及时、准确、全方位的新闻消息

 1
1 2
2 3
3 4
4 5
5 6
6 7
7 8
8 9
9 10
10 1
1 2
2 3
3 4
4 5
5 6
6 7
7 8
8 9
9 10
10








